This Dr. Axe content is medically reviewed or fact checked to ensure factually accurate information.
With strict editorial sourcing guidelines, we only link to academic research institutions, reputable media sites and, when research is available, medically peer-reviewed studies. Note that the numbers in parentheses (1, 2, etc.) are clickable links to these studies.
The information in our articles is NOT intended to replace a one-on-one relationship with a qualified health care professional and is not intended as medical advice.
This article is based on scientific evidence, written by experts and fact checked by our trained editorial staff. Note that the numbers in parentheses (1, 2, etc.) are clickable links to medically peer-reviewed studies.
Our team includes licensed nutritionists and dietitians, certified health education specialists, as well as certified strength and conditioning specialists, personal trainers and corrective exercise specialists. Our team aims to be not only thorough with its research, but also objective and unbiased.
The information in our articles is NOT intended to replace a one-on-one relationship with a qualified health care professional and is not intended as medical advice.
5 Ways to Naturally Manage Huntington’s Disease
December 6, 2015
You’ve most likely heard of disorders like ALS, Parkinson’s disease and Alzheimer’s disease, but you may not have heard about an equally tragic condition called Huntington’s disease (HD).
Capable of damaging nerves and disturbing important chemical-signaling processes that take place between the brain and other body parts, some people even describe common Huntington’s disease symptoms as “like having ALS, Parkinson’s and Alzheimer’s simultaneously.”
Clearly, this is a condition that can drastically affect someone from going about even normal, everyday activities. Unfortunately, there is no known cure — however, research suggests supplementation may help manage some symptoms of Huntington’s disease.
So what are the signs and symptoms of Huntington’s disease, and what treatment options are out there to help curb and potentially reverse this debilitating condition? Let’s explore.
What Is Huntington’s Disease?
Huntington’s disease is an unfortunate and somewhat rare genetic brain disorder that currently affects about 30,000 Americans. According to the Huntington’s Disease Society of America, HD is called a “family disease” because children who have a parent with HD have about a 50/50 chance of carrying the faulty gene themselves. (1)
Sadly, people with HD usually experience drastic personality changes, loss of memory and impaired motor skills over the course of 10–25 years, which makes it hard to live a normal, functioning life.
HD is a disabling disease that disturbs someone’s ability to think, reason, connect socially, remember information and move. While emerging research suggests that certain natural supplements might be able to help stall the progression of HD, currently it’s classified as a progressive fatal disorder that has no known proven cure.
Despite the option to perform testing to find out if someone has inherited the HD gene, according to survey results published by Harvard University and the MassGeneral Institute for Neurodegenerative Disease:
More than 90 percent of those in the United States who know they are at risk for HD because of their family history have abstained from genetic testing, often because they fear discrimination or don’t want to face the stress and anxiety of knowing they are destined to develop such a devastating disease. (2)
Conventional Treatment for Huntington’s Disease
Traditionally, most physicians prescribe a number of medications to help control the various emotional and physical symptoms of HD, although these are used to make living easier and aren’t yet able address the underlying problem at its root.
As of 2008, some progress was made when the U.S. Food and Drug Administration approved the drug tetrabenazine to treat involuntary writhing movements of HD (chorea), making it the very first Huntington’s disease drug approved for use in the U.S.
In 2017, an experimental drug was introduced in a human trial involving 46 patients with early Huntington’s disease. The human trial began in late 2015 and utilized the drug, IONIS-HTTRx, which was injected into the spinal fluid for it to reach the patient’s brain. The results of the trial confirmed the drug lowered the level of the toxic disease-provoking protein, huntingtin. (3)
The trial also highlighted that the drug was well-tolerated; however, vital long-term data is still needed to confirm if lowering the levels of huntingtin will change this disease’s course and if the disease can ultimately be prevented before symptoms develop. Although more data and trials are needed, animal research showed some motor function was recovered in those experiments and suggests this experimental drug could change the course of Huntington’s disease. The breakthrough with this research and trial could present further opportunities for other neurodegenerative diseases. (4)
As of early 2018, the study has not been published in a journal and is still active. (5)
Other conventional treatments for HD include antidepressants (for mood swings and depression), mood stabilizers, antipsychotics and/or benzodiazepines (for involuntary movement, secondary to tetrabenazine).
One of the downsides to the conventional treatment for Huntington’s disease and other cognitive disorders is that they commonly have a lot of side effects, such as fatigue, insomnia, appetite and mood changes, etc. (6)
6 Natural Ways to Manage Huntington’s Disease
A somewhat effective treatment plan for Huntington’s disease may be holistic — one that treats the “whole person” with cognitive skill building, supplements, an anti-inflammatory diet and appropriate physical activity.
What else besides medications might be able to help reduce symptoms of Huntington’s disease? Here are several ways that practitioners are now managing HD symptoms:
1. Reduce Inflammation
Whether it’s a disorder of the cardiovascular, endocrine, immune or central nervous system, inflammation only makes matters worse. Scientific investigations find that high inflammation levels and oxidative stress caused by free radicals can speed up neurodegenerative disease progression and worsen symptoms. (7)
Using electronic and other technologies, researchers are now beginning to better understand how inflammation is related to mutated cells, defective energy metabolism (a defect in the mitochondria) and oxidative stress (normal metabolic activity in the brain that produces toxic compounds called free radicals), which all contribute to disease formation.
Inflammation, worsened by factors like a poor diet, environmental pollution, toxin exposure, high stress levels and inactivity, can affect immunity and the body’s “tropic factors,” meaning the natural chemical substances that are supposed to protect against cell changes and death. (8)
To lower inflammation, it’s important to eat a healing diet that’s nutrient-dense, limit use of harsh chemicals in household/beauty products, avoid smoking, stay active and try to manage stress.
2. Maintain Physical Activity
It’s been found that people with HD can greatly benefit from staying active as long as they can. Doctors consider it “extremely important for people with HD to maintain physical fitness as much as possible” since those who exercise and keep active tend to maintain better control over physical movements for longer. (9)
While exercise or even everyday activity can become harder as time goes on, regular movement and exercise can make a big impact. With neurodegenerative diseases, it’s also now accepted that regular and sustained physical activity has the potential to benefit cardiovascular health and other factors that can impair quality of life and lead to complications, adding this to the list of the benefits of exercise.
Studies have shown that in HD patients, physical activity can help manage stress from social stigma, lack of motivation and trouble with executive functions. (10)
There are also multiple methods of in-home exercise regimens that have been studied to be effective in small study pools, from exercise DVDs to supervised Dance Dance Revolution™ sessions! (11, 12)
3. Adjust Diet to Stall Weight Loss
Throughout the progression of Huntington’s disease, weight loss usually occurs, sometimes very quickly and to the point that it causes serious complications. While it becomes harder and harder to chew normally and safely, changing someone’s diet to make sure he or she consumes enough nutrients and calories is important. (13)
This helps fight complications of being underweight like worsened depression, very low energy, thyroid gland changes and poor digestion. Helping people with HD maintain their appetites as long as possible can be very helpful. It’s also useful to make food easier to consume, such as pureeing or blending foods into smoothies, soups, etc.
In addition, intermittent fasting and the keto diet have been shown to have positive effects on neurological disorders, so it may not hurt to try these options under the supervision of a doctor. (14) In fact, more than one animal study has discovered a potential benefits of the ketogenic diet or intermittent fasting in delaying weight loss, managing glucose and protecting neurons from injury. (15, 16)

4. Cognitive Training
Setting up a clear schedule, following a routine and practicing reminders of daily life seem to be helpful when it comes to managing cognitive and psychiatric disorders. Doctors recommend that family and caregivers of those with HD help create an environment that limits stress, too much difficult decision-making and the need to learn new information often. This can mean: (13)
- Using calendars and clear schedules
- Creating a predictable routine
- Setting reminders
- Keeping the living area organized
- Prioritizing certain activities over others
- Staying social and practicing hobbies to lower stress
- Breaking down difficult tasks into manageable steps
- Creating a calm living environment that is structured and has limited uncertainty
- Avoiding family conflict, fights and other stressors
5. Natural Supplementation
Early studies done by Massachusetts General Hospital in 2004 found that high doses of certain supplements, namely the nutritional compound called creatine can help delay the onset of symptoms of Huntington’s disease. Creatine was safe and well-tolerated by most of the 64 study participants who were either confirmed to be carrying the HD gene or at a high risk but not yet tested. Using neuroimaging studies, the researchers found that treatment slowed regional brain atrophy and progression of presymptomatic HD. (17)
HD damages brain cells by interfering with cellular energy production, leading to a depletion of adenosine triphosphate (ATP). ATP is the underlying molecule that powers most biological processes and essentially gives our cells”energy.” Creatine has long been known to help restore ATP and maintain cellular energy, which is why it’s being studied in the treatment of Parkinson’s disease, Huntington’s, ALS and spinal cord injuries, which are all impacted by neurodegeneration. (18)
At least one review of available evidence states, “Current literature suggests that exogenous creatine supplementation is most efficacious as a treatment paradigm in Huntington’s and Parkinson’s disease but appears to be less effective for ALS and Alzheimer’s disease.” (19)
In the past, privacy and patient autonomy have been an obstacle in testing genetic disorders. The 2014 study design was one of the first of its kind to test genetic diseases without needing subjects to first be screened for whether or not they carried the gene, since some preferred not to know. (17)
Professors of neurology at Harvard Medical School continued to study the effects of creatine in HD patients by leading a worldwide phase 3 trial (CREST-E) of high-dose creatine in early symptomatic HD. Unfortunately, their results found that placebo actually outperformed high-dose creatine, which led them to reverse their earlier hypothesis. (20)
This doesn’t necessarily mean that creatine is a pointless option, but it didn’t seem to be effective for slowing functional decline in patients with early symptomatic Huntington’s. Further research still needs to be conducted to find out if the effects may be related more to the prevention of initial symptoms or if it is more effective at some point during the progression of the disease.
In addition, it’s important to note that the dosage used in these studies is at such a high level that it shouldn’t be taken by anyone without close medical supervision.
6. Physical and Occupational Therapy
In addition to exercise, physical therapy seems to be a potentially beneficial treatment method for some symptoms of Huntington’s disease. A 2002 case study of a 49-year-old male with HD recognized significant improvements in disability markers after 14 weeks of an in-home physical therapy exercise program. This led the authors to believe more research was needed on this connection. (21)
Then, in 2008, researchers at Cardiff University in the UK conducted questionnaires and interviews with 49 physical therapists working with HD patients. Their results led them to realize that physical therapy is still very under-used for these patients (especially in early-stage Huntington’s), the benchmarks of success weren’t well-defined and that the main “treatment aim” of these therapists is to successfully use physical therapy to decrease falls and mobility deficits. Subsequently, the authors of this study created a “conceptual framework for physical therapy intervention in HD” based on their findings. They believe their framework may be used in complex neurodegenerative disorders, including Huntington’s, and might help to inform future trials on the impact of physical therapy on HD symptoms. (22)
A small-scale study with twelve patients diagnosed with Huntington’s disease found that physical therapy over a six-week period improved gait issues and determined more defined methods for assessing their results. (23)
Occupational therapy, focused on adapting to normal life skills even with reduced physical function, has also helped some Huntington’s patients improve their quality of life. A 2007 pilot study of “intensive rehabilitation,” including breathing exercises, speech therapy, physical therapy, occupational therapy and cognitive rehabilitation exercises found no decline in motor or cognitive decline over the two years of the study. (24) While it’s difficult to attribute these results to one individual method, as no controls were used, it’s still significant, as a two-year period for a person with HD is almost always marked by traceable decline in motor skills and cognition.
Unfortunately, these methods are very underused by HD patients. One survey conducted found that only eight percent of Huntington’s patients had been seen by a physical therapist, 24 percent by an occupational therapist and close to zero by a speech therapist. (25)
On the Horizon: More Hope for Treating Huntington’s Disease
Aside from creatine, numerous other supplements and complimentary forms of medicine are being studied (mostly in animal trials, some in small human trials) in regard to their ability to slow, prevent or manage symptoms brain and nerve damage. Some include: (26)
- Resveratrol (27, 28, 29, 30, 31)
- Coenzyme Q10 (CoQ10) (32, 33, 34, 35, 36, 37)
- Vitamin E (26, 37)
- Ethyl-EPA (38, 39, 40, 41, 42, 43)
- Idebenone (26, 44)
- Unsaturated fatty acids (45)
Results from various studies using supplements/herbs have been mixed so far, with some patients experiencing improvements and others not reaching statistical significance that suggests they’re getting better (I have included both positive and negative results above). (46) Some reasons for this may include variations in sources and dosages of each natural remedy, study design and even the possibility that the studied method truly isn’t effective when it is tested widely.
While there is still a long road to travel, there are some hopeful bits of progress that may eventually lead to exciting discoveries.
Additional lifestyle modifications or methods are now commonly suggested to manage cognitive disorders and support overall brain health, although they may or may not have any specific impact on Huntington’s disease. Examples of these include:
- avoiding chronic stress
- focusing on individualizing treatments and treating the whole person
- promoting relaxation, self-care and self-healing
- focusing on good nutrition and a nutrient-dense diet that is anti-inflammatory, particularly full of healthy fats
- using preventive practices like exercise, sleep and avoiding toxin exposure
- the ketogenic diet
- stem cell therapy
- brain-supporting essential oils such as rosemary, frankincense and turmeric oil
- lion’s mane mushroom
While we need to wait to see what research emerges in years to come, it makes sense that even for genetic disorders, helping people stay in the best of health overall — including physically, mentally, spiritually and emotionally — will likely give them the best chance of a satisfying life.
Symptoms of Huntington’s Disease
Like certain other cognitive or nerve disorders, Huntington’s disease symptoms aren’t usually present from a young age. Most people start developing HD symptoms between the ages of 30 and 50. Once they begin, symptoms tend to worsen over the next one to two decades until the disorder reaches a fatal point.
HD patients become very weak and as their immune systems suffer, which results in many people eventually developing illnesses like pneumonia or heart complications. While normally a healthy person can overcome these obstacles, someone with Huntington’s disease isn’t capable of recovering.
Common symptoms of Huntington’s disease include: (47)
- Personality changes and mood disturbances
- Symptoms of depression
- Mood swings
- Memory loss, forgetfulness
- Impaired judgment and reasoning
- Slurred speech
- Involuntary movements (known as chorea)
- Difficulty swallowing and eating
- Loss of appetite, significant weight loss
How Huntington’s Disease Develops and Progresses
HD manifests by affecting nerves spread throughout just about the entire brain, including the striatum, subthalamic nucleus and substancia nigra. Certain areas of the brain are more vulnerable to the effects of nerve damage than others. The area called the basal ganglia is where a group of nerves cells are clustered together, which are called nuclei. HD damages parts of the nuelei, which are responsible for regulating body movements and also behaviors.
The “control center” of the brain is another area that’s damaged by HD, which is why someone’s judgment, rationalization and moods are also negatively impacted. Degeneration in this areas is what leads people over time to feel like they’re “losing their minds” with age.
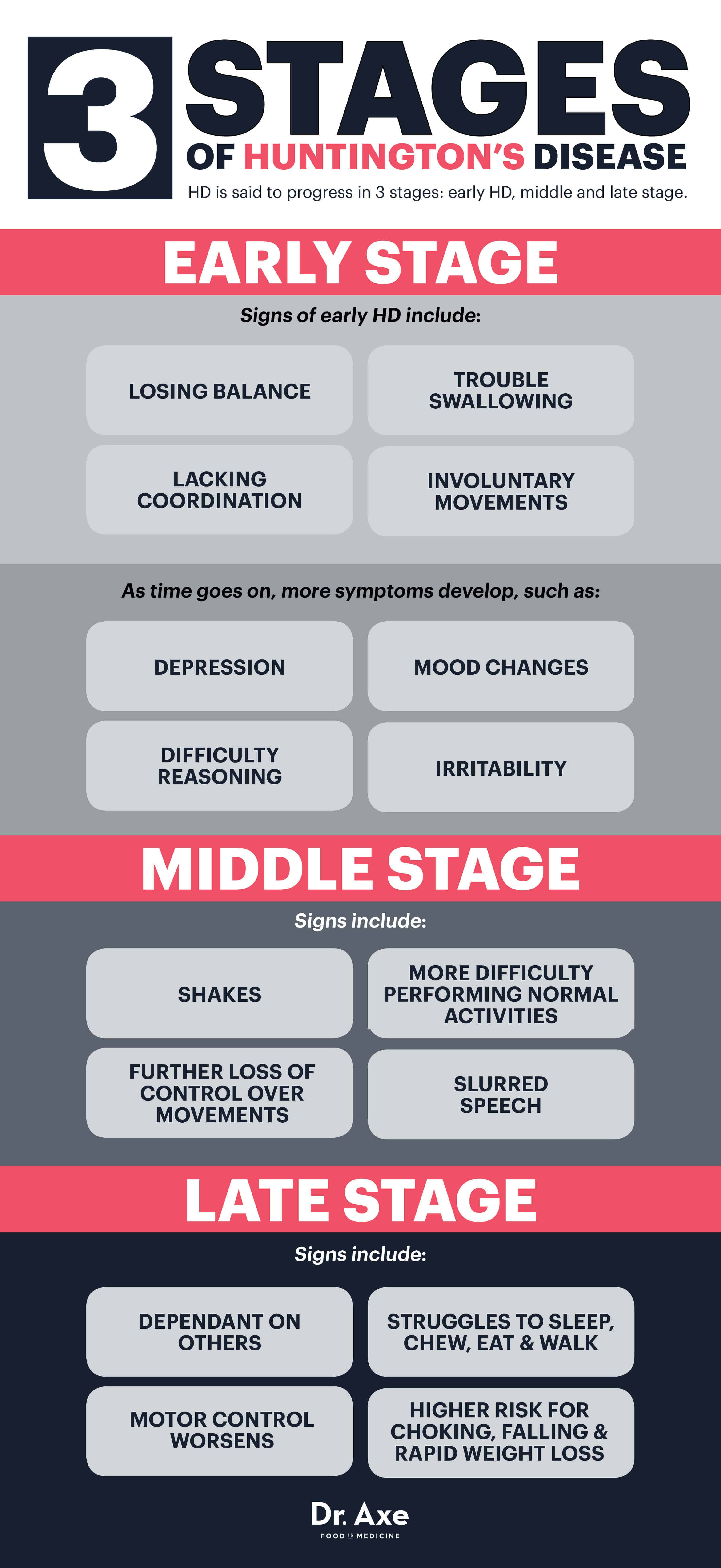
HD is said to progress in three stages: early HD, middle and late stage. (48)
Early Stage Huntington’s Disease
At first, someone experiences only subtle, sometimes unnoticeable symptoms like losing their balance, lacking coordination, or having some trouble swallowing and controlling the tongue. Others can start to see signs of involuntary movements (chorea) while in the early stages of HD.
As time goes on, difficulty reasoning and mood changes continue to develop. Someone might become depressed, irritable or prone to mood fluctuations — which can partially be due to the changes happening in the brain, but also worsened once a Huntington’s disease diagnoses is made. At this point, some doctors prescribe mood-controlling medications to help alleviate symptoms of depression, but as HD worsens, it becomes more and more difficult to work, maintain relationships and live alone.
Middle Stage Huntington’s Disease
Middle stage HD results in a further loss of physical control over movements, as nerves continue to become damaged. Ordinary activities and leading a normal life become harder to do at this point. Involuntary movements, shakes and slurred speech are common, which people often associate with being characteristic of other disorders like MS or Parkinson’s disease.
Medications can sometimes help the lack of movement control (chorea), while occupational and physical therapists might also enter the equation to further assist with coordination, stability, swallowing and walking.
Late Stage Huntington’s Disease
By the time someone has had HD for a number of years, he or she is usually totally dependent on others and might live in a full-time facility. Motor control becomes worsened in most cases, while someone also struggles to speak, chew, eat and walk. The parts of the brain responsible for memory, language and comprehending information also continue to decline, which means it’s hard to put sentences together or remember other people.
Because it’s hard to control the tongue and swallow normally, choking is also a major concern at this point, which means HD patients cannot eat alone. Once HD becomes fatal, it’s not the actual disorder that causes a person to die, but rather the illnesses they acquire in the process of its progression (like infections or heart problems) or complications of the disease (like choking, falling and rapid weight loss).
Causes of Huntington’s Disease and How It’s Passed On
Surprisingly, we all carry a certain gene that ‘s linked to Huntington’s disease — however, people who wind up developing the disorder must inherit another specific genetic factor that expands and worsens the disorder. The “expanded” HD gene is passed from parent to child, and every person who inherits the gene eventually develops the disease. Whether one child in a family inherits the gene or not has no effect on whether other children in the family will or will not. (49)
While roughly 30,000 Americans have a diagnoses of HD, another 200,000 are at risk for developing the disorder genetically but have not yet started to display symptoms. (1) Both men and women are equally susceptible to developing HD, and it affects people of all nationalities, ethnic groups and religions throughout the world.
Huntington’s disease is genetic (offspring inheriting an affected gene have a 50 percent chance of the disorder progressing), inherited and considered “autosomal dominant.” This means that the likelihood of the expanded HD gene passing from parent to child does not depend on the child’s gender; both females and males have an equal chance of being affected.
One of the saddest things about Huntington’s disease is that it often devastates entire families, since it can affect family members for many generations and make it hard to sustain relationships or a positive outlook. Children of patients with HD usually face a very high level of stress due to both the uncertainty of having the disorder potentially develop in years to come, plus the responsibility of taking care of a sick parent.
One of the hardest decisions a family usually faces is whether or not to perform prenatal or genetic testing in order to know if an unborn baby or child is affected by carrying the HD gene. Because there currently isn’t a proven cure or prevention method, there isn’t necessarily anything positive or preventative people can do once they find out they carry the gene or have passed it on to their offspring. However, some people choose to do testing as early as possible in order to know what’s ahead or to have more choices about how to handle the future.
In about 10 percent of cases, HD symptoms emerge in children or teens, since the vast majority don’t show symptoms until years later during middle age. This is called juvenile Huntington’s disease (JHD). Symptoms of JHD are somewhat different than adult onset HD and tend to get worse faster than adult HD. Common symptoms that appear in children can include trouble walking, instability, clumsiness or changes in speech.
Before the age of 18, genetic testing for HD is prohibited because authorities fear that children will not understand the full implications of HD or benefit from knowing what’s in their futures. If children begin showing signs of HD at a very young age before they turn 18, tests can be performed to confirm a partial diagnoses. If a woman is pregnant and wants to find out if the baby carries the gene, she can receive testing for the fetus early on between weeks 10–18 of her pregnancy.
Takeaways on Huntington’s Disease
While dealing with Huntington’s disease can feel hopeless at times, there are preventative measures and natural treatments to slow and even perhaps reverse this dreadful disease. There may not be a cure, but maintaining a healthy, organic diet is a key to reducing inflammation and naturally managing symptoms of cognitive disorders like HD.
In addition, cognitive training along with physical activity promote brain health, which can strengthen the defense against rapid cognitive decline. And of course, there is promising research on the use of supplements to curb and potentially reverse this debilitating disorder.